Research Focus
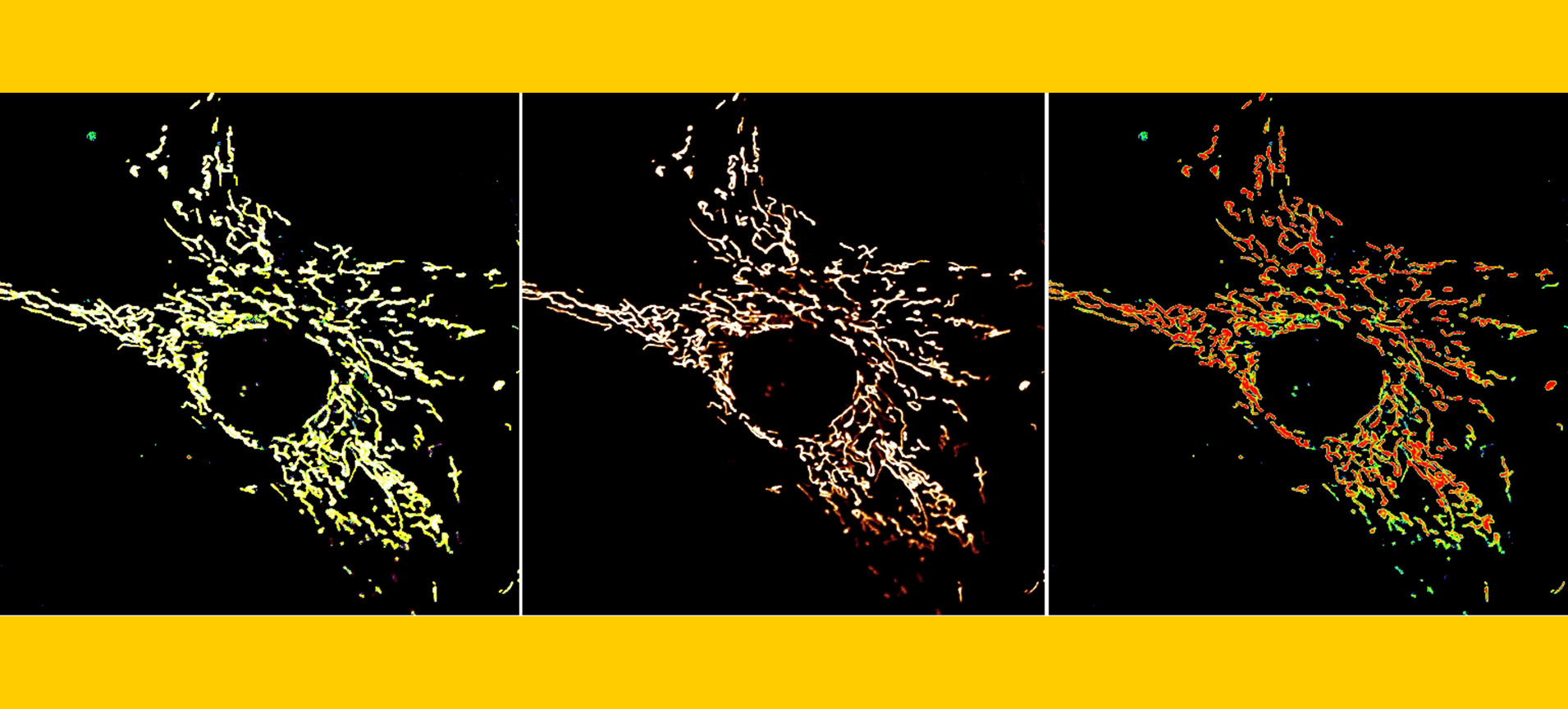
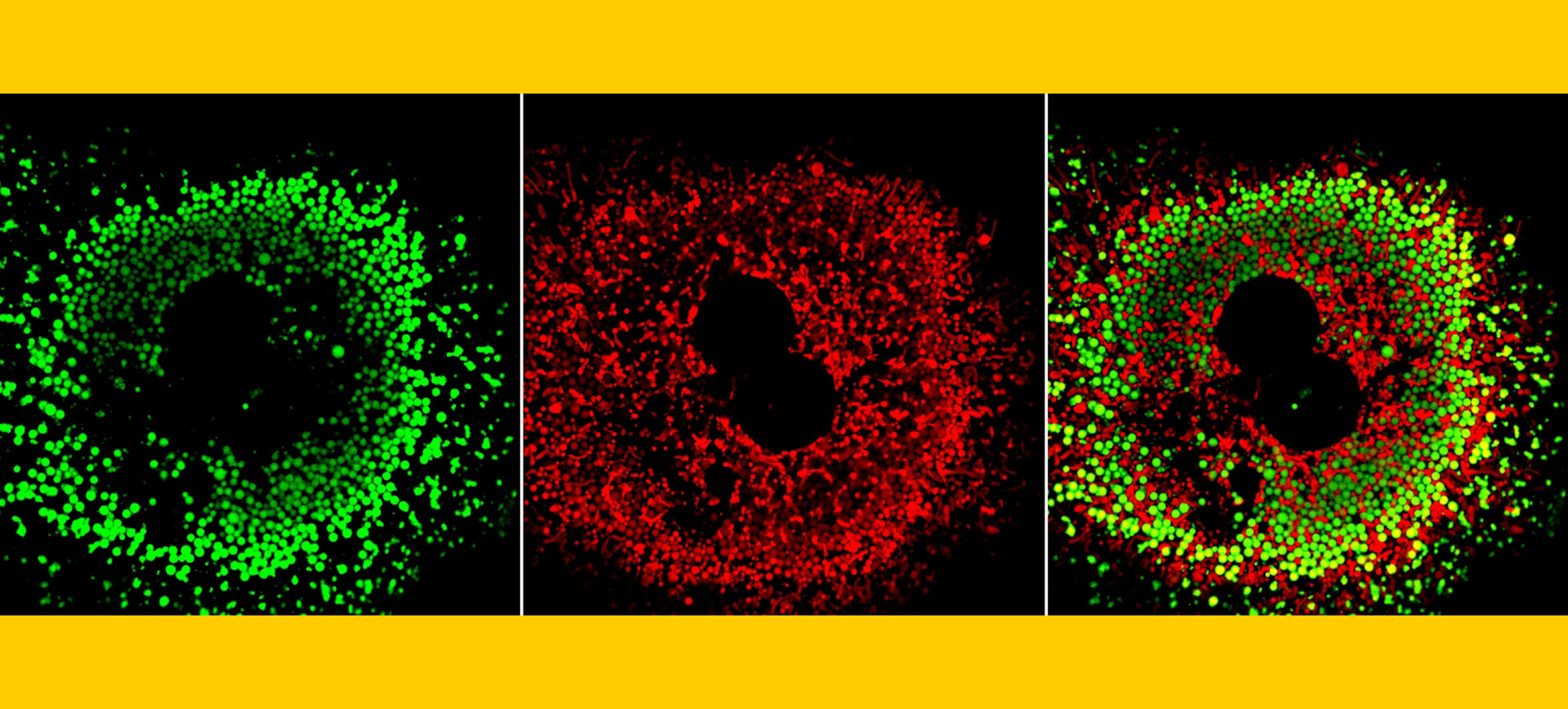
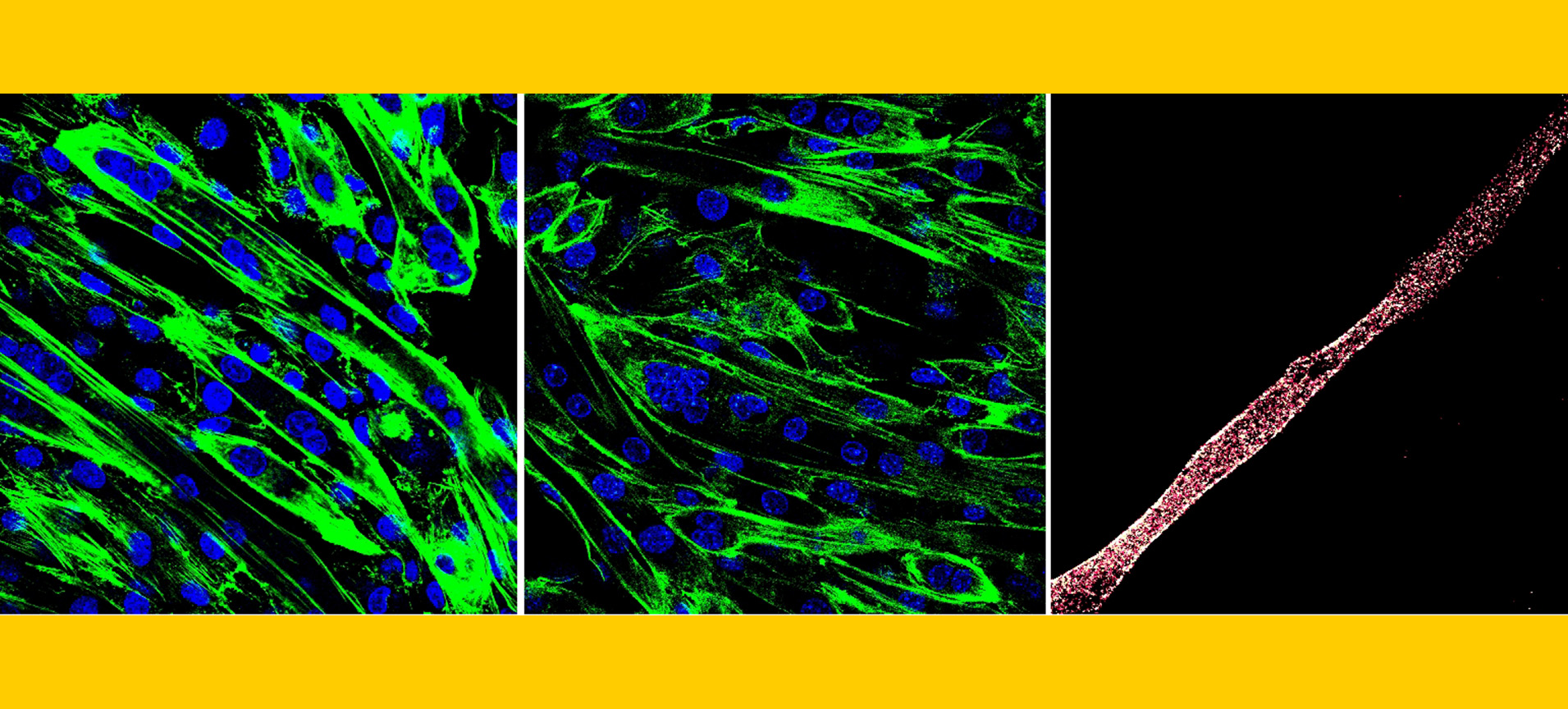
Research Focus
Research Focus
The primary focus of our laboratory is the study of mitochondria, which are ancient endomembrane systems that originated approximately two billion years ago through the engulfment of a proteobacterium by a precursor of modern eukaryotic cells. Mitochondria possess an intrinsic capability to generate ATP via respiration, establishing them as a crucial driving force in the process of evolution. In addition to their well-recognized role as the cell’s powerhouse, mitochondria have acquired numerous other functions, including the capacity for calcium buffering, the generation of reactive oxygen species, the regulation of apoptosis, activation of the endoplasmic reticulum stress response, and the extensive implications of mitochondrial dysfunction. Mitochondria are implicated in a variety of prevalent diseases, including, but not limited to, Alzheimer’s disease, Parkinson’s disease, metabolic disorders, muscular dystrophy, cardiovascular diseases, and also play a significant role in the normal aging process. Our research employs biochemical physiology in conjunction with animal models to systematically investigate the role of mitochondrial ion signaling in both health and disease. Our long-term objective is to elucidate the underlying molecular mechanisms associated with these diseases and to identify and develop potential therapeutic interventions.
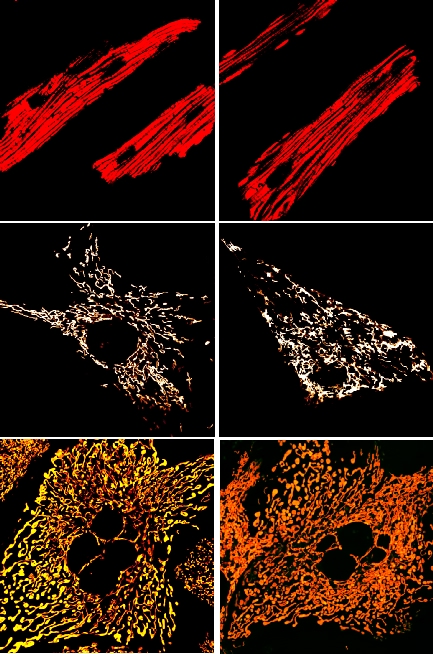
Mighty Magnesium
Magnesium is a crucial intracellular divalent cation that serves as a cofactor in various genomic processes, including replication and transcription. It is compartmentalized in the mitochondria, with Mrs2 being the main entity for magnesium influx. Besides activating enzymes in the TCA cycle and electron transport chain, our research has shown that mitochondrial magnesium binds to the N-terminal domain of the mitochondrial calcium Uniporter (MCU), regulating calcium uptake (Cell Chemical Biology, 2016). We created a conditional tissue-specific Mrs2 knockout mouse model using the CRISPR/Cas9 technique to explore how magnesium levels affect mitochondrial calcium uptake. Our findings indicate that disrupted magnesium uptake leads to mitochondrial calcium overload, bioenergetic crises, and the opening of the mitochondrial permeability transition pore (Mitochondrion, 2024). Nonetheless, the precise mechanisms driving MCU-mediated calcium overload during ischemia/reperfusion injury remain unclear, particularly given that Mrs2 systematically regulates MCU channel activity. With NIH R01 funding, we aim to elucidate how MCU escapes magnesium-dependent regulation, leading to increased calcium overload in disease.
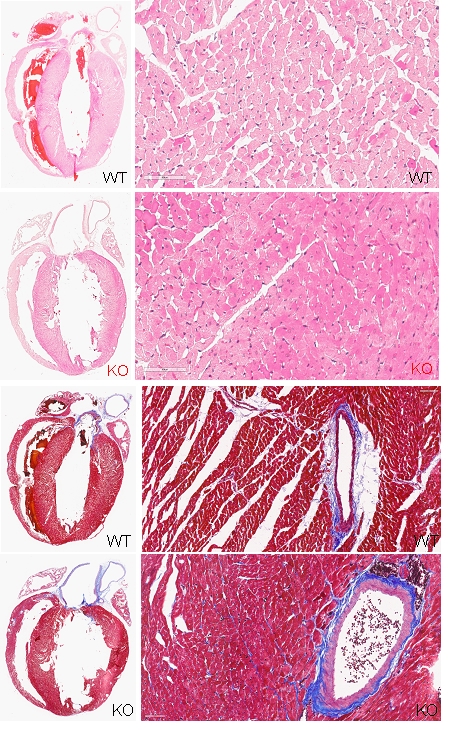
Mitochondrial calcium and McArdle disease: Exploring potential solutions for cardiac issues.
Myophosphorylase deficiency, known as McArdle disease (Glycogen Storage Disease type V), is the most common metabolic myopathy, inherited in an autosomal recessive pattern due to mutations in the PYGM gene. This results in a deficiency of the myophosphorylase enzyme, crucial for glycogen breakdown in skeletal muscles. While McArdle disease typically does not cause multisystem complications, some cases have reported cardiac issues. The condition lacks the skeletal muscle isoenzyme, though human cardiac tissue contains three isoenzymes. Recent studies indicate that despite sufficient myocardial myophosphorylase, patients may experience cardiac complications such as electrocardiographic abnormalities and heart failure. This suggests a link between McArdle disease and cardiac dysfunction, potentially exacerbated by mitochondrial calcium uptake leading to bioenergetic disruptions. We aim to investigate how mitochondrial calcium uptake is altered in the cardiac tissues of individuals with McArdle disease to understand its role in metabolic disorders.

Mitochondrial Magnesium and Hepatic Lipid Metabolism.
Magnesium serves as a crucial cofactor in lipid metabolism. They modulate the activity of LCAT and LPL, both of which contribute to reducing TG levels and elevating HDL cholesterol levels. Furthermore, magnesium functions as a regulatory element on 3-HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. It has been proposed that dysregulation of magnesium homeostasis may hinder the inactivation of HMG-CoA reductase through phosphorylation mechanisms. Emphasizing the importance of maintaining magnesium homeostasis in lipid metabolism. Studies using mouse models have demonstrated that magnesium dysregulation leads to increased TG synthesis in the liver, resulting in hypertriglyceridemia, elevated levels of free cholesterol, and decreased concentrations of esterified cholesterol. Subsequent investigations have linked magnesium dysregulation with a greater incidence of insulin resistance, obesity, and hypertension. Moreover, such dysregulation can exacerbate and hasten lipid deposition and the formation of lesions in the coronary arteries. Characteristically, magnesium dysregulation manifests as elevated concentrations of chylomicrons, VLDL, and LDL, accompanied by reduced HDL levels. Despite these findings, the intricate relationship between mitochondrial magnesium in maintaining lipid metabolism remains inadequately understood and necessitates further exploration. We aim to determine whether disturbances in mitochondrial magnesium homeostasis affect hepatic lipid metabolism.
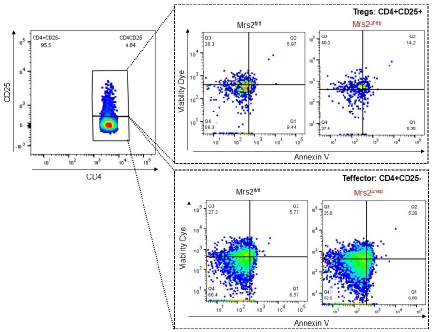
Mitochondrial Magnesium and Treg health.
Regulatory T cells (Treg cells) play a crucial role in immune regulation, preventing the immune system from attacking the body’s own tissues and thereby maintaining homeostasis. Recent studies suggest that magnesium intake may enhance the quantity and functionality of Treg cells, which could benefit individuals with autoimmune disorders. Magnesium supplementation has been linked to an increase in Foxp3+ Treg cells, known for their immune-suppressing capabilities, potentially reducing the severity of autoimmune diseases. This effect of magnesium on Treg cell activity may help alleviate inflammation and improve health outcomes. Our goal is to explore how mitochondrial magnesium affects Treg cells in specific health contexts.